Molecular Dynamics of the Silica-Water Interface
Explore ab initio MD simulations of the silica-water interface. Learn about Density Functional Theory (DFT), atom density profiles, and H-bond analysis.
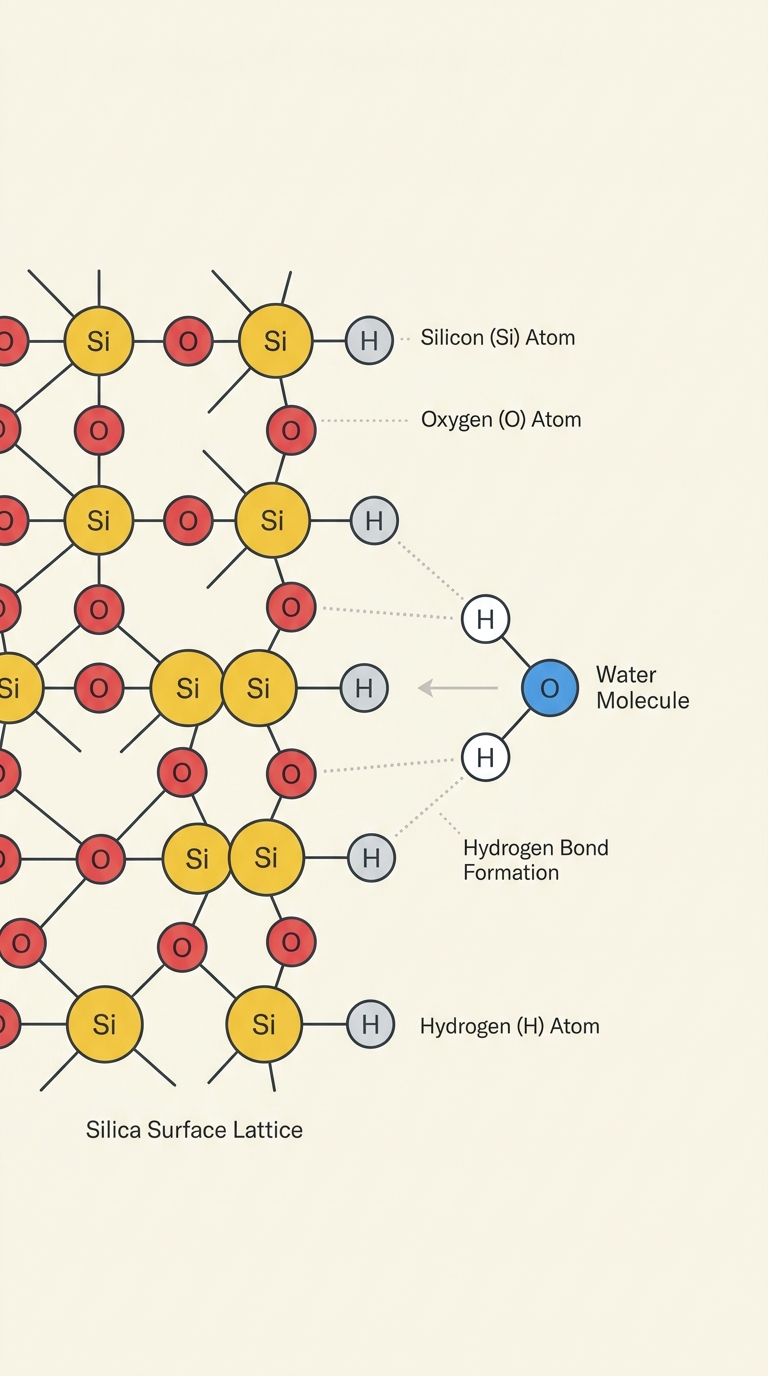
The Silica-Water Interface
from the Analysis of Molecular Dynamic Simulations
Sheikha Faisal Lardhi
Master of Science Thesis
King Abdullah University of Science and Technology (KAUST)
Thuwal, Kingdom of Saudi Arabia · May 2013
Research Overview & Objectives
Core Question:
How does liquid water interact with a silica surface at the molecular level?
Investigate the silica-water interface microscopically using ab initio MD simulations
Calculate atomic density profiles of water layers near the β-cristobalite surface
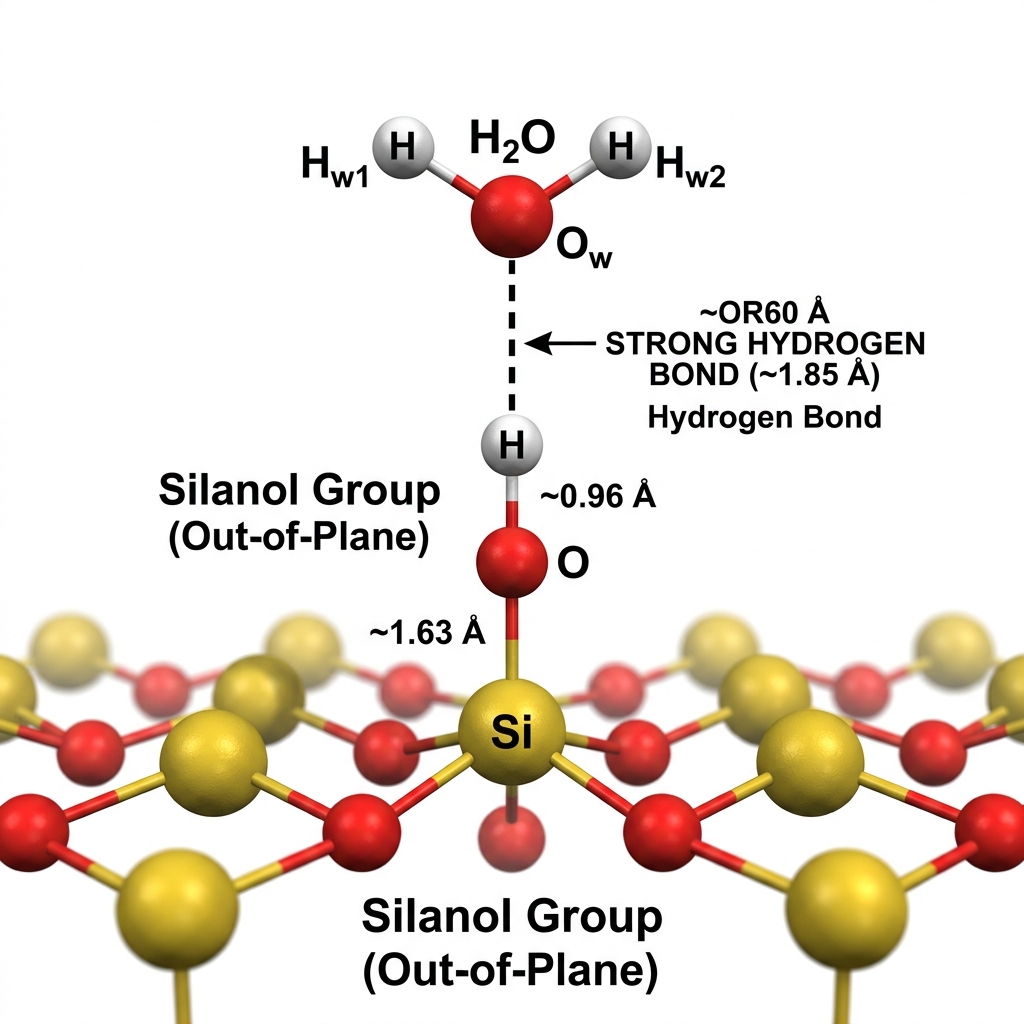
Analyze radial distribution functions (RDF) of hydrogen bonds at surface silanols
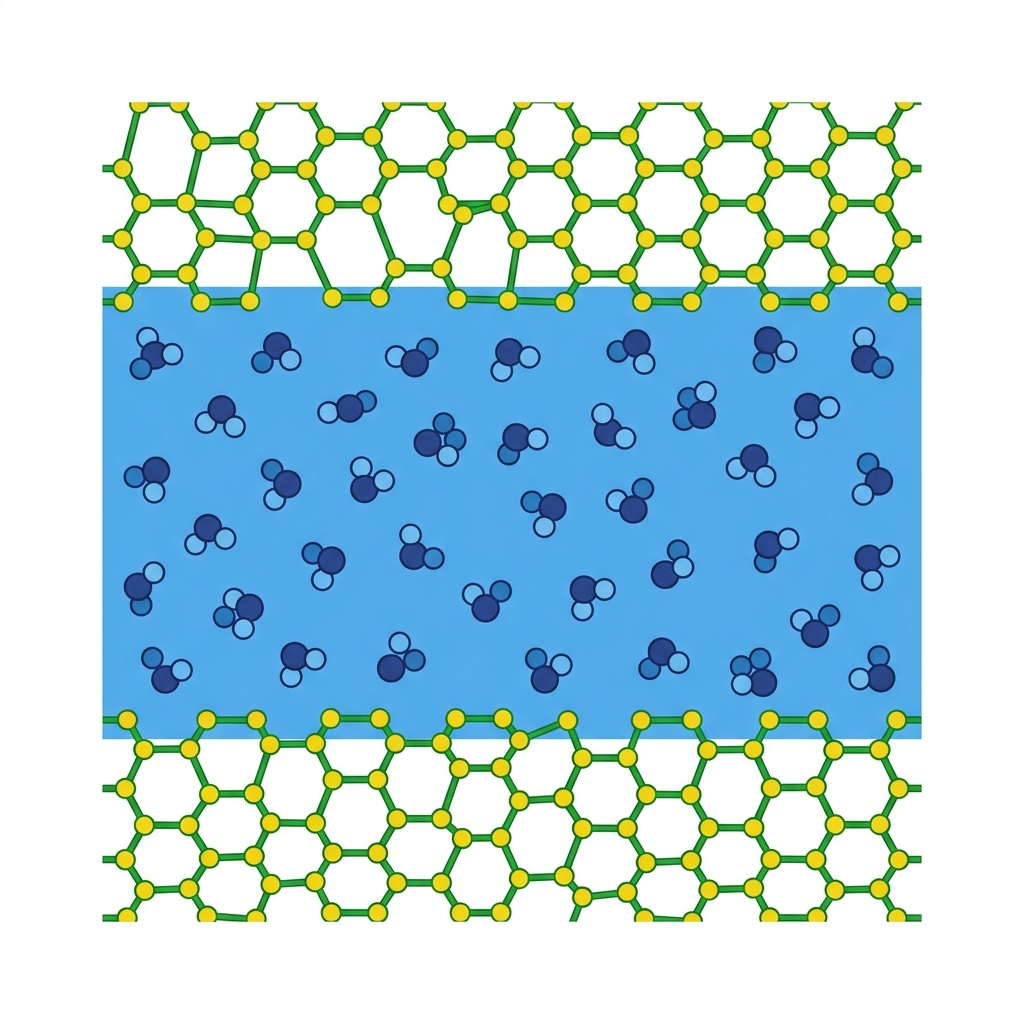
System: bulk liquid water confined between two β-cristobalite silica surfaces · CP2K ab initio MD · 25 picoseconds
Sheikha Faisal Lardhi · KAUST 2013
2
Molecular Dynamics: Principles & Applications
Computational Chemistry Group
3
First applied by Alder & Wainwright, 1950s
1 million atom simulation of Satellite Tobacco Mosaic Virus (STMV), 2006 — 50 ns using NAMD
Initial Positions & Velocities
Calculate Forces (Newton's Laws)
Integrate Equations of Motion
New Positions & Trajectory
Structural Biology
(protein folding)
(thin films, nanotech)
(silica-water)
(bond breaking)
Biophysics
(NMR refinement)
The Silica-Water Interface: Background & Importance
Theoretical Framework: DFT & Electronic Structure
From QM to DFT
Schrödinger Equation: Hψ = Eψ
describes full quantum state of the system
Born-Oppenheimer Approximation
nuclei fixed; electrons move in nuclear field (mass ratio electron:proton = 1:1836)
Density Functional Theory (DFT)
energy expressed as a functional of electron density ρ(r), not wavefunctions
HK Theorem 1
V<sub>ext</sub> uniquely defined by ρ(r)
HK Theorem 2
Variational principle — approximate ρ always gives E ≥ E<sub>0</sub>
Exchange-Correlation Functionals
Functional
Type
Used For
PBE
GGA
This study (fast, robust)
BLYP
GGA
Hydrogen bond analysis
B3LYP
Hybrid
Higher accuracy
This study uses:
PBE functional
+ DZVP-GTH-BLYP basis set + GTH pseudo-potentials
Slide 5
Computational Methodology: CP2K & Quickstep
CP2K Program
(Fortran 95, GPL, 900,000+ lines)
Quickstep DFT Module
GPW Approach
Gaussian + Plane Waves
MD Trajectory Generation
Run on 64 cores
CRESCO supercomputer
Intel Xeon 5160 @ 3.00 GHz
10 Teraflops
Basis Sets
STOs — accurate but computationally expensive
GTOs — linear combination of Gaussian primitives
DZVP: Double-Zeta Valence Polarization
Split-valence sets balance accuracy vs. cost
Pseudo Potentials (GTH)
Replace inner-shell electrons with analytical functions
Reduces computational cost significantly
Includes relativistic effects
Essential for transition metals & large systems
Ab initio MD cost:
~100x more expensive than classical MD per water molecule
SLIDE 6
Model Construction & Simulation Setup
Structure A — Sparse Water
96 atoms total | 10 SiO₂ units (5 layers) | 18 H₂O molecules
Box: a=b=7.34Å, c=20.0Å
Min O-Si distance: 4.0 Å
Structure B — Dense Water
117 atoms total | 10 SiO₂ units (5 layers) | 25 H₂O molecules
Same box dimensions
Min O-Si distance: 3.5 Å
Geometry Optimization
BFGS, 2000 iterations, 24 cores, ~1 hour
Equilibration
NVT ensemble, 300K, 5×5ps = 25ps total, 0.5 fs timestep
Trajectory Analysis
Density profile + RDF
Periodic Boundary Conditions (PBC) applied — simulates infinite bulk system
7
Results: Atom Density Profiles
Water Layering Near Silica Surface
Water organizes in well-structured distinct layers
Structure A:
water NOT strongly adsorbed at surface
Structure B:
strong adsorption at 2Å — denser water fills silanol spaces
Both show symmetric distribution — confirmed equilibration
Slide 8
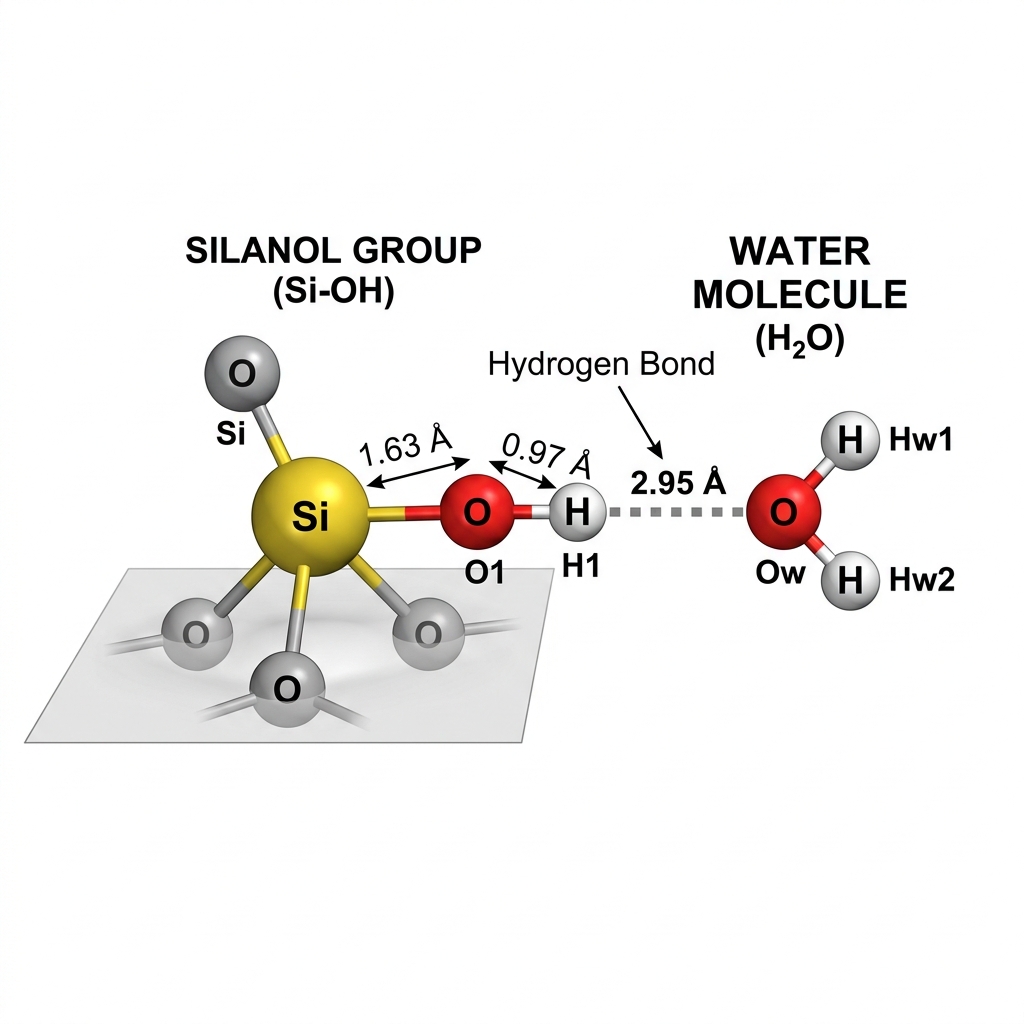
Results: Radial Distribution Function (RDF) & H-Bonding
What does g(r) tell us?
Slide 9
Summary & Future Directions
Summary of Findings
Simulation Protocol ✓
Built & optimized two silica-water structures (A: sparse, B: dense) using CP2K ab initio MD at 300K for 25 ps
Density Profiles ✓
Water forms well-structured layers; Structure B shows stronger surface adsorption at ~2Å with molecules intercalating silanol groups
RDF Analysis ✓
Strong H-bond network confirmed; peaks at 1.5–2.0Å for out-of-plane silanols; water density does not dramatically alter H-bond structure
Future Directions
Other SiO₂ polymorphs: α-quartz, α-cristobalite
Gold nanoparticle–water interfaces
Graphene–water interfaces
Metal oxides for photocatalytic water splitting
Supervised by Prof. Luigi Cavallo · Committee: Prof. David Keyes & Prof. Mikhail Moshkov · KAUST CEMSE & KCC · 2013
Thank You
Slide 10
- molecular-dynamics
- silica-water-interface
- computational-chemistry
- dft
- ab-initio-md
- surface-science
- nanotechnology