








Structure-Based Drug Discovery: SBDD Principles & Methods
Expert guide to Structure-Based Drug Discovery (SBDD), covering molecular docking, virtual screening, X-ray crystallography, and ADMET optimization.
Structure-Based
Drug Discovery
Decoding Molecular Architecture to Design Tomorrow's Medicines
Advanced Course in Medicinal Chemistry & Computational Biology
01 / 30
Academic Presentation | 2026
Table of Contents
Introduction to Drug Discovery
Fundamentals of Protein Structure
Target Identification & Validation
Structural Determination Methods (X-ray, NMR, Cryo-EM)
Molecular Docking & Virtual Screening
Fragment-Based Drug Discovery
Lead Optimization & SAR
Computational Tools & AI in SBDD
Case Studies & Success Stories
Challenges & Future Perspectives
02 / 30
Section 1: Introduction to Drug Discovery
From Disease Biology to Therapeutic Molecules
03 / 30
Academic Presentation | 2026
What is
Drug Discovery?
The rigorous, multi-disciplinary process of finding new candidate medications, moving from identifying initial disease targets to establishing safe and effective treatments.
Target Identification & Validation
Discovering specific biological mechanisms linked to disease pathology.
Hit to Lead & Lead Optimization
Screening vast compound libraries and chemically refining the best candidates.
Preclinical & Clinical Trials
Testing in laboratory models initially, followed by safely testing in human volunteers.
Regulatory Approval
Detailed review by health authorities to secure marketing authorization.
10,000
Compounds Screened
250
Preclinical Candidates
5
Clinical Trials
1
Approved Drug
04 / 30
Academic Presentation | 2026
Traditional
vs.
Structure-Based
Drug Discovery
Traditional / Phenotypic
Trial-and-error screening
Blind to molecular mechanisms
High attrition rates
Slow and expensive
Early aspirin discovery
Structure-Based (SBDD)
Rational, target-guided design
Uses 3D protein structures
Higher hit rates
Faster lead optimization
HIV protease inhibitors
SBDD reduces development costs by up to 50% and significantly improves clinical success rates.
05 / 30
HISTORY & MILESTONES OF
STRUCTURE-BASED DRUG DISCOVERY
1970s
First protein X-ray crystal structures solved
1980s
HIV crisis drives SBDD urgency
1994
FDA approves first SBDD drug (Saquinavir)
1996
Indinavir & Ritonavir trigger SBDD revolution
2001
Imatinib (Gleevec) cancer kinase inhibitor
2006
Cryo-EM revolution begins
2012
GPCR structures solved; new drug targets
2020s
AI/ML accelerates SBDD (AlphaFold breakthrough)
06 / 30
Academic Presentation | 2026
02
Fundamentals of Protein Structure
The Molecular Targets at the Heart of SBDD
07 / 30
Academic Presentation | 2026
Levels of Protein Structure
PRIMARY
Amino acid sequence; linear polypeptide chain; defined by genetic code
SECONDARY
α-helices and β-sheets; hydrogen bond stabilized; local folding motifs
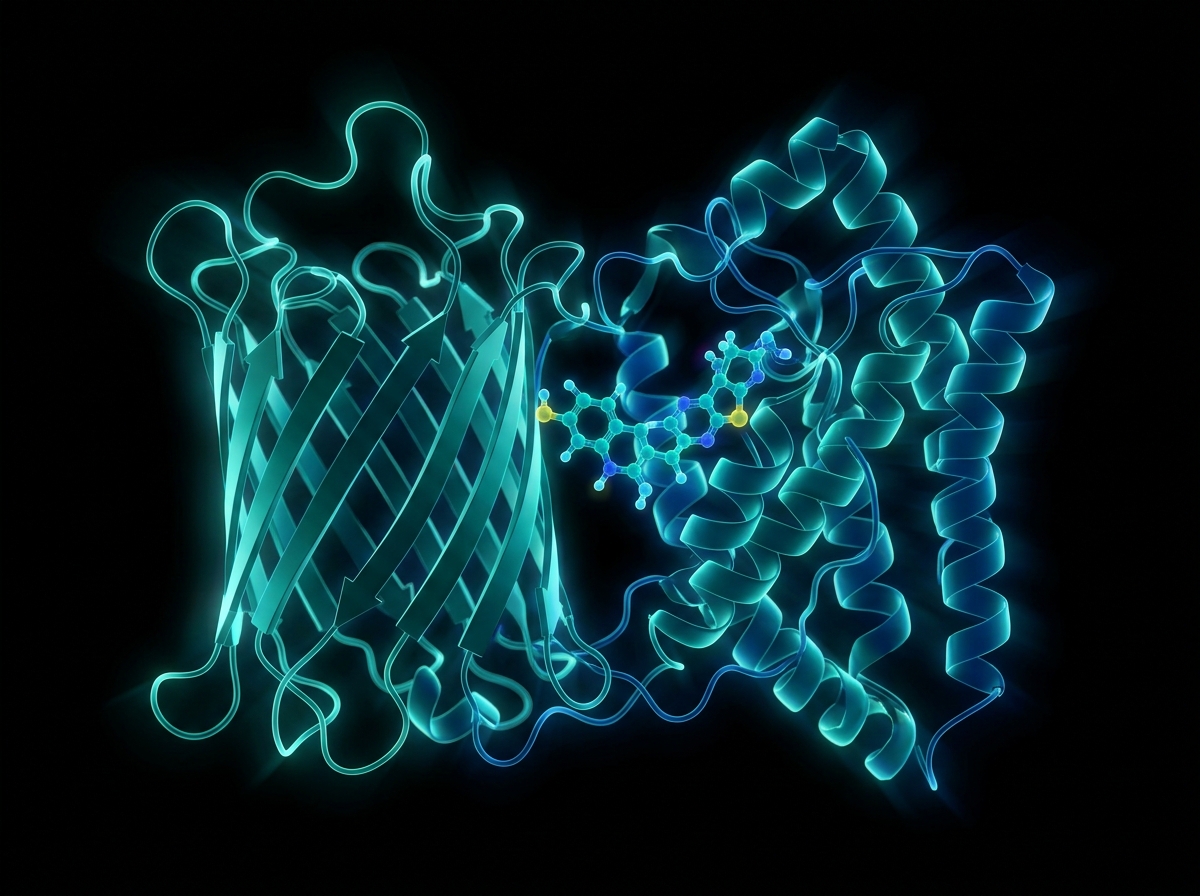
TERTIARY
3D folded conformation of a single polypeptide; hydrophobic core; binding pockets formed here
QUATERNARY
Assembly of multiple polypeptide subunits; oligomeric complexes; allosteric sites
Binding pockets emerge at the tertiary/quaternary level — these are the targets for SBDD.
08 / 30
Binding Sites
& Pharmacophores
A specific 3D region of a protein with complementary shape and chemical properties to a ligand
Orthosteric (active) site
Allosteric site
Cryptic/hidden sites
Protein-protein interaction interfaces
Shape complementarity
Electrostatics
Hydrophobic contacts
H-bond donors/acceptors
09 / 30
Advanced Medicinal Chemistry
03
Target Identification <br/>& Validation
Finding the Right Biological Lock for Our Chemical Key
10 / 30
Major Drug Target Classes
ENZYMES
Kinases, proteases, phosphatases; inhibit catalytic activity; ~47% of all drug targets
GPCRs
G-protein coupled receptors; ~34% of FDA-approved drugs; 7-transmembrane helices
ION CHANNELS
Voltage/ligand-gated channels; CNS, cardiac targets
NUCLEAR RECEPTORS
Transcription factor regulation; steroid hormones, cancer
PROTEIN-PROTEIN INTERACTIONS
Challenging; large flat surfaces; emerging frontier
NUCLEIC ACIDS
DNA/RNA targeting; antivirals, oncology
11 / 30
Target Validation Strategies
Genetic Validation
Knockout/knockin models, CRISPR screens, human genetics (GWAS)
Biochemical Validation
In vitro binding assays, enzymatic activity, SPR, ITC
Cell-Based Validation
Knockdown/overexpression, phenotypic rescue experiments
In Vivo Validation
Animal models, disease-relevant endpoints
Clinical Biomarker
Target engagement confirmed in patients
Key Validation Criteria
Druggability
Disease Linkage
Selectivity Potential
Structural Data Availability
Druggable
Disease-Relevant
Structurally Characterized
Good Target
12 / 30
Target Validation Overview
04
Structural Determination Methods
Visualizing Proteins at Atomic Resolution
13 / 30
X-ray Crystallography
Gold standard for protein structure determination since the 1950s
Protein expression & purification
Crystal growth
(key bottleneck)
X-ray diffraction data collection (synchrotron)
Phase determination (MR, SAD, MAD)
Model building & refinement
Deposition to Protein Data Bank (PDB)
High resolution (1-3 Å), well-established, large structures possible
Requires crystallization, static snapshots, difficult for membrane proteins
14 / 30
Cryo-EM and NMR Spectroscopy
Cryo-EM
No crystallization needed
Maintains near-native conditions
Awarded Nobel Prize in 2017
Ideal for large macromolecular complexes
Achieves sub-2 Ångström resolution
NMR Spectroscopy
Measures solution state dynamics
Optimal for small proteins (< 50 kDa)
Reveals conformational flexibility
Yields detailed chemical shift data
15 / 30
05
Molecular Docking & Virtual Screening
Computational Prediction of Ligand-Target Interactions
16 / 30
Academic Presentation | 2026
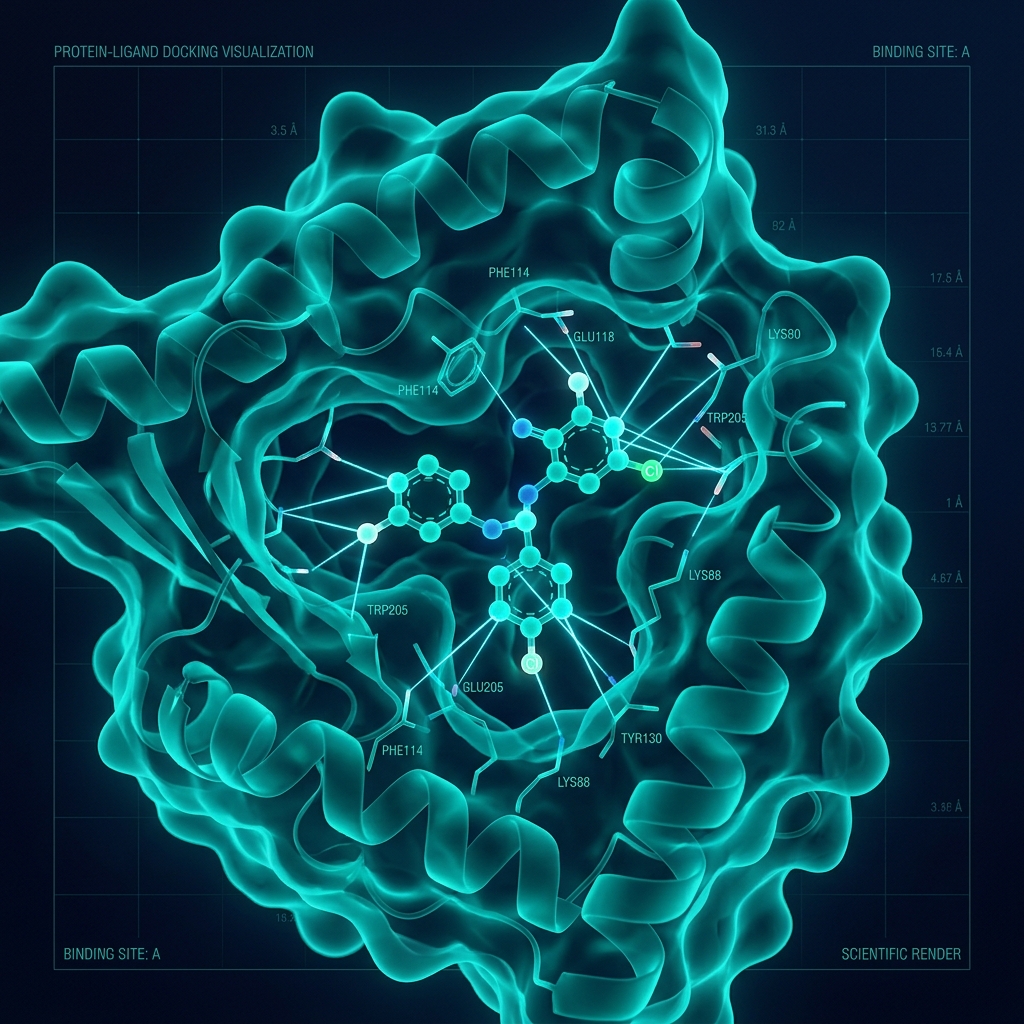
What is docking?
Computational method predicting preferred orientation and binding affinity of a small molecule within a protein binding site
Two Main Components
SEARCH ALGORITHM
explores conformational space (rigid, semi-flexible, fully flexible)
Genetic algorithms, Monte Carlo, systematic search
SCORING FUNCTION
ranks poses by predicted binding affinity
Force-field based, empirical, knowledge-based, ML-based
AutoDock, Glide, GOLD, Vina, rDock
Protein flexibility, solvation, entropy, scoring accuracy
17 / 30
Virtual Screening Workflow
Compound Library
~10M+
Druglikeness Filter
~1M
Pharmacophore Screening
~100K
Molecular Docking
~10K
Re-scoring
~1K
Experimental Validation
5-20 hits
18 / 30
Academic Presentation | 2026
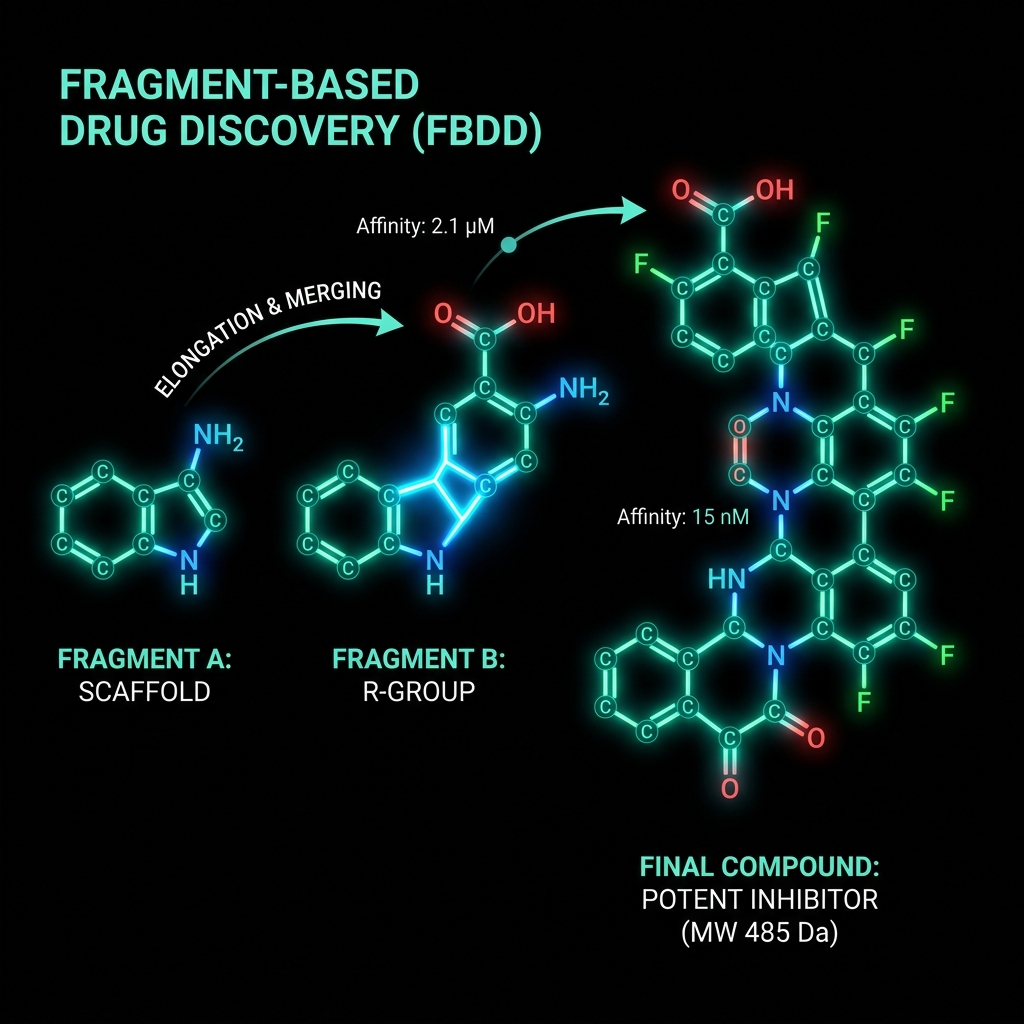
Fragment-Based Drug Discovery (FBDD)
Building Potent Drugs from Small Molecular Fragments
19 / 30
Fragment-Based Drug Discovery (FBDD)
FBDD uses very small molecules (150-300 Da) as starting points, identified by weak but efficient binding, then grown or linked into drug-like leads.
Better chemical space coverage
Higher ligand efficiency
Fewer compounds to screen (~1,000-10,000 vs millions)
20 / 30
07
Lead Optimization &
Structure-Activity Relationships
Fine-Tuning Molecules for Potency, Selectivity & Safety
21 / 30
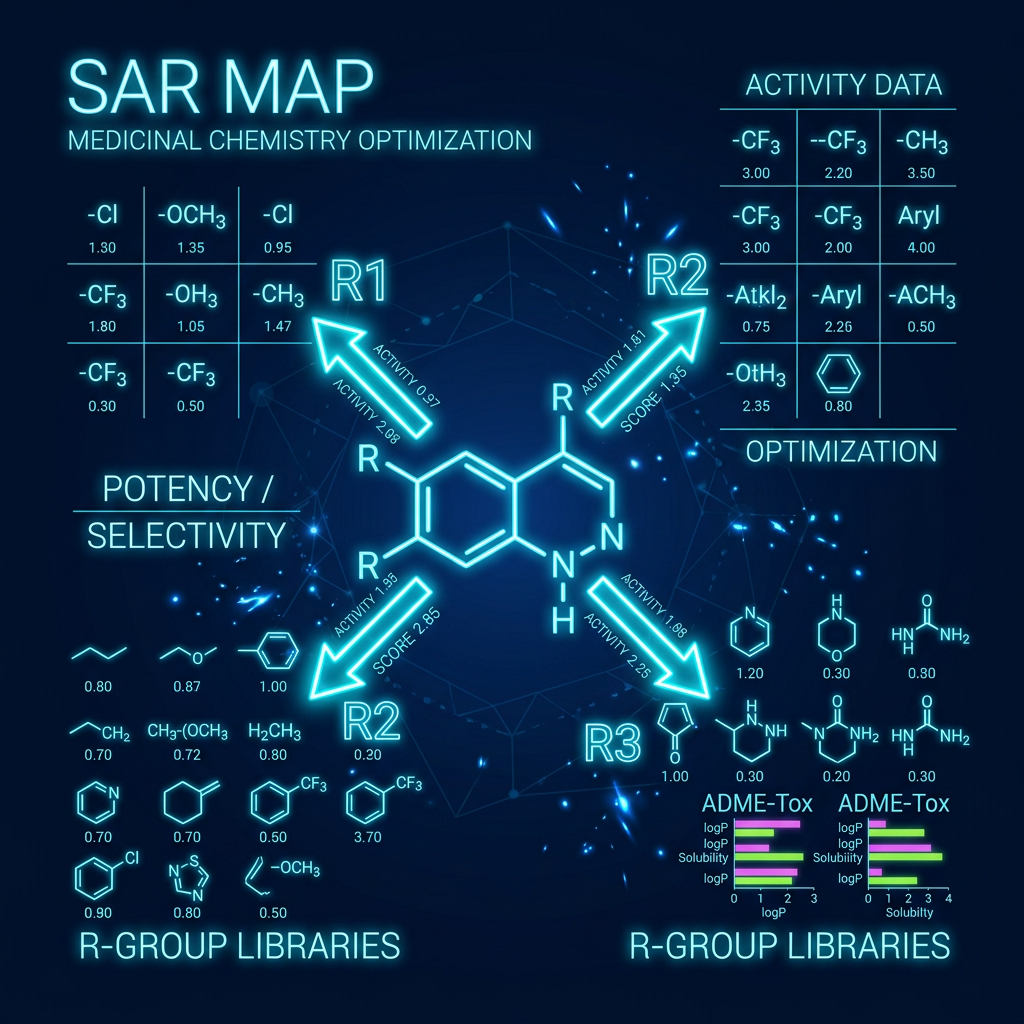
Structure-Activity Relationships (SAR)
SAR describes how structural modifications to a molecule affect its biological activity at a target.
Bioisosteric replacements
maintain activity while improving ADMET
Scaffold hopping
change core while retaining pharmacophore
Matched molecular pairs (MMP) analysis
Free-Wilson analysis
Activity cliffs
small structural change causes large potency jump
22 / 30
Academic Presentation | 2026
40%
of all drug candidate failures in clinical trials are directly attributed to poor ADMET properties.
23 / 30
Academic Presentation | 2026
- drug-discovery
- medicinal-chemistry
- molecular-docking
- sbdd
- computational-biology
- biotechnology
- pharmaceuticals